At a glance
- The "Long" in IGF-1 LR3 describes a 13-amino-acid peptide extension, not a long half-life. That naming trips up most buyers.
- Both analogs dodge IGF binding proteins; Francis et al. (1992) ranked Long[Arg3]-IGF-I and des(1-3)IGF-I as the two most potent variants, roughly 2.5-fold above native IGF-I in living tissue.
- IGF-1 DES has a very short half-life (minutes) and drives the unproven "local/site-specific" injection theory. No human trial supports it.
- Both cross-react with the insulin receptor and can cause hypoglycemia; IGF-1 lowers glucose even in mice with no insulin receptors (Di Cola et al. 1997).
- Zero human performance trials exist for either compound. Everything below is preclinical or in-vitro.
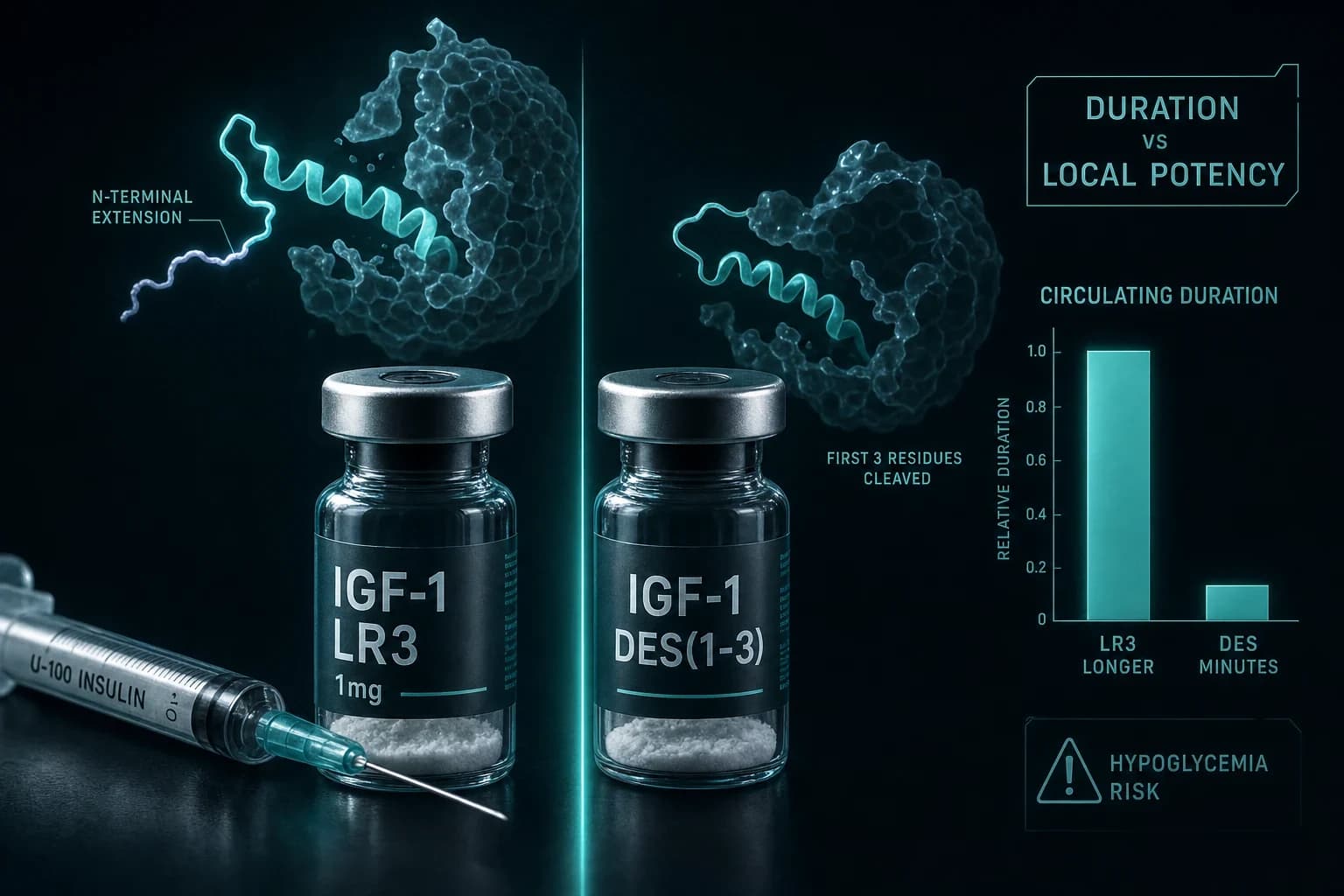
Long R3 IGF-1 does not have a long half-life because it is "Long." The word describes 13 extra amino acids bolted onto the front of the peptide, not how long the molecule lingers in your blood. That one misread powers most of the bad advice about how IGF-1 LR3 and IGF-1 DES actually differ.
Both are engineered to solve the same problem: native IGF-1 gets grabbed by IGF binding proteins the moment it hits circulation, and a peptide handcuffed to a binding protein cannot reach its receptor. LR3 and DES take two different routes around that fence. LR3 rebuilds the front of the molecule. DES amputates it. The result is two analogs that share a goal and almost nothing else in how they behave.
Here is the part nobody selling these will lead with. Neither compound has been through a single human performance trial. Everything that follows comes from cell culture, rodents, and one antidoping detection study. Read it that way.
Why native IGF-1 needs an escort in the first place
More than 99% of the IGF-1 circulating in your body is not free. It rides around bound to a family of six IGF binding proteins, mostly IGFBP-3, in a large ternary complex with a protein called ALS. That complex is a storage depot. It keeps IGF-1 from flooding the insulin receptor and crashing your blood sugar, and it slowly releases the hormone where tissues need it.
The N-terminal end of the IGF-1 molecule, the first few residues, does a lot of the binding-protein contact. Change that region and you change how tightly IGFBPs can hold on. That is the entire design premise behind both analogs. Weaken the binding-protein grip, and more of the peptide stays free to hit the type 1 IGF receptor (IGF-1R) that drives muscle protein synthesis and cell proliferation.
The catch is that the binding proteins are also what give native IGF-1 its reservoir. Strip them away and you get a more potent molecule with, in theory, less of a buffer against the insulin-like glucose crash. Hold that thought for the hypoglycemia section.
Two ways to dodge a binding protein
IGF-1 LR3 is native human IGF-1 with two modifications. First, a 13-amino-acid extension peptide (derived from the N-terminus of methionyl porcine growth hormone) is fused to the front. Second, the glutamic acid normally at position 3 is swapped for arginine, which is where "R3" comes from. Together these knock IGFBP affinity down to roughly 2 to 4% of native, while receptor binding stays close to normal.
IGF-1 DES, full name des(1-3)IGF-I, goes the other direction. Instead of adding, it subtracts: the first three residues (Gly-Pro-Glu) are cleaved off. Losing the glutamate at position 3 removes a key binding-protein contact point, so DES also binds IGFBPs far more weakly than native IGF-1, with reported reductions ranging from about 25-fold up to two orders of magnitude depending on which binding protein and assay you look at. DES is not purely synthetic exotica, either. It occurs naturally in bovine colostrum, human brain tissue, and porcine uterus, where proteases clip native IGF-1 down to the truncated form (Ballard et al. (1996)).
So both weaken the same brake. The interesting question is what that actually buys you in living tissue, and that is where the one genuinely good structure-activity paper comes in.
What Francis actually measured in 1992
The foundational data here is Francis et al. (1992) in the Journal of Molecular Endocrinology. They built a panel of IGF-I analogs, including Long[Arg3]-IGF-I and des(1-3)IGF-I, and tested them in two kinds of cells: ones that secrete IGF binding proteins and ones that do not.
In binding-protein-rich cells, the potency rank order was Long[Arg3]-IGF-I equal to des(1-3)IGF-I, both greater than Long[Gly3]-IGF-I, greater than Long IGF-I, greater than plain IGF-I. The two IGFBP-evaders sat at the top, together.
Then the reveal. In fibroblasts that do not secrete binding proteins, Long[Arg3]-IGF-I became less potent than native IGF-I. Read that twice. The whole advantage of LR3 evaporated, and then inverted, the moment there were no binding proteins to evade. Francis and colleagues concluded that the enhanced potency comes from altered IGFBP interaction, not from any improvement in receptor binding. The analogs are not better keys. They are keys that skip the coat check.
That finding reframes the entire LR3-versus-DES debate. Neither analog is intrinsically stronger at the receptor. They are stronger only in the presence of the binding proteins they were built to escape.
The comparison, side by side
| Feature | IGF-1 LR3 | IGF-1 DES(1-3) |
|---|---|---|
| Structure | Native IGF-I plus a 13-residue N-terminal extension, plus Arg substituted at position 3 | Native IGF-I with the first 3 residues (Gly-Pro-Glu) removed |
| IGFBP binding | ~2-4% of native affinity | Roughly 25-fold to 100-fold lower than native, depending on binding protein |
| IGF-1R (receptor) binding | Near-native; ~3-fold lower in some assays | Near-native |
| Half-life (commonly cited) | ~20-30 h, systemic (weakly sourced) | Minutes to ~20-30 min, very short |
| In-tissue potency | ~2.5-fold over native in catabolic rats | ~2.5-fold over native in rats; ~10-fold in some cell assays |
| Hypothesized use | Whole-body, "always on" signaling | Local injection into a target muscle |
| Human performance evidence | None | None |
Half-life is where the marketing gets slippery
The clean story sold online is: LR3 is the long-acting systemic one, DES is the short-acting local one. It is not that clean.
Start with the rodent anabolic data. Tomas et al. (1992) infused native IGF-I, des(1-3)IGF-I, and LR3IGF-I into dexamethasone-treated rats, a catabolic model. Both analogs came in about 2.5-fold more potent than native IGF-I for nitrogen retention and weight gain, and this held even though LR3 bound the type 1 receptor about 3-fold worse than native. Again, the edge traced back to escaping the binding proteins, not to receptor performance. Note the number: 2.5-fold, not the 10-fold or "insane" figures thrown around in forums. That larger figure comes from certain cell assays, not from whole animals.
Now the half-life claim itself. The frequently repeated "20 to 30 hours" for LR3 does not come from a clean human pharmacokinetic study, because none exists. The one rigorous look at these specific molecules in vivo is an antidoping paper, Mongongu et al. (2021), which tracked LongR3-IGF-I, des(1-3)-IGF-I, and R3-IGF-I after intramuscular injection in rats using mass spectrometry. Their detection windows scramble the tidy narrative: intact des(1-3)-IGF-I stayed detectable for around 24 hours, while intact LongR3-IGF-I disappeared within about 4 hours, with its degradation products persisting longer. That is a detection window at an injection site, not a plasma half-life, but it should make you distrust any confident single-number half-life you read on a product page. Mongongu's team also flagged what they were really studying: black-market bodybuilding products, never approved for human use, frequently contaminated with oxidized, degraded peptide.
Bottom line: The "Long" in LR3 is about peptide length, not duration of action, and the popular half-life numbers for both analogs rest on vendor lore rather than human data. Treat "20-30 hours" as a claim, not a fact.
The local-potency hypothesis behind DES
DES's short half-life is the whole basis of its reputation. The bodybuilding theory goes like this: because des(1-3)IGF-I is cleared so fast, if you inject it directly into a trained muscle, it will fire the local IGF-1 receptors right there before it washes out and reaches the rest of the body. Site-specific growth. Bigger biceps on the arm you inject, in the fantasy version.
There is a sliver of biological plausibility. A very short-lived, potent, binding-protein-independent agonist injected into tissue will hit that tissue first. But "plausible" and "demonstrated" are different words, and the second one does not apply here. There is no human study showing localized, injection-site-specific hypertrophy from DES. The idea that you can meaningfully partition IGF-1 signaling to one muscle by manipulating half-life is a hypothesis that has never cleared a controlled trial in people.
It is also worth being precise about potency. Ballard et al. (1996) describe des(1-3)IGF-I as about 10-fold more potent than IGF-I at stimulating hypertrophy and proliferation of cultured cells, all of it downstream of reduced binding-protein binding. That 10-fold figure is real, and it is a cell-culture number. In whole animals the gap compressed to roughly 2.5-fold. In-vitro potency is a ceiling, not a promise.
And even the "IGFBP-evading" premise has limits. Ricort & Binoux (2001) showed that IGFBP-3 still suppressed des(1-3)IGF-I signaling: 2.5 nM IGFBP-3 knocked out more than 50% of the stimulation from 3 nM des(1-3)IGF-I, and 10 nM cut it by more than 80%, through a receptor-level mechanism independent of ligand binding. DES dodges the binding proteins. It does not become invisible to them.
The hypoglycemia problem nobody markets
Here is the risk that both compounds share, and it is not a footnote. IGF-1 and insulin are structural cousins, roughly 50% homologous, and their receptors cross-react. IGF-1 can bind and activate the insulin receptor, and it lowers blood glucose.
How real is the crossover? Di Cola et al. (1997) gave IGF-1 to mice engineered with no insulin receptors at all. It still caused a prompt, sustained drop in plasma glucose, essentially identical to the response in normal mice, by driving muscle glucose uptake and suppressing hepatic glucose output through the IGF-1 receptor itself. Translation: you do not even need the insulin receptor for IGF-1 to crash your blood sugar. Its own receptor does the job.
Now layer the analogs on top. LR3 and DES are built to stay free rather than bound in the IGFBP reservoir that normally buffers native IGF-1's insulin-like activity. A more available, more potent IGF-1R agonist with no binding-protein brake is exactly the profile you would design if you wanted an unpredictable glucose drop.
Warning: Both IGF-1 LR3 and IGF-1 DES can cause hypoglycemia, and it can be abrupt. The IGFBP-evading design that makes them potent is the same feature that removes the natural buffer against a glucose crash. This is a genuine hazard, not a talking point, and it is one more reason these compounds sit outside any legitimate self-experimentation.
Where these sit next to the growth-hormone-axis options
People cross-shop these against secretagogues, which is a category error worth naming. MK-677 (ibutamoren) and CJC-1295 work upstream: they nudge your own pituitary to release more growth hormone, which then raises your own IGF-1 through the liver, inside the intact IGFBP system, with your body's own feedback loops still in charge. LR3 and DES skip all of that and hammer the IGF-1 receptor directly, downstream of every regulatory checkpoint the secretagogue route preserves.
That is a fundamentally different risk posture. A GH secretagogue that overshoots still runs into negative feedback and the binding-protein buffer. A direct IGF-1R agonist with the buffer engineered out does not have those guardrails. It is the difference between turning up a thermostat and pouring the raw signal straight into the wire.
If you are reconstituting research peptides of any kind, get the math right before anything else; the reconstitution calculator exists so a decimal slip does not become a dosing error, which matters far more when the molecule in the vial can drop your blood glucose.
The honest scorecard
Strip out the forum confidence and here is what the literature actually supports. Both analogs are more potent than native IGF-1 in the presence of binding proteins, roughly 2.5-fold in whole animals, because they escape the IGFBP handcuffs rather than because they bind the receptor better (Francis 1992, Tomas 1992). DES is more potent still in cell culture, around 10-fold, and is cleared fast, which is the seed of the unproven local-injection theory (Ballard 1996). Neither has a clean human half-life number. Neither has a human performance trial. Both can cause hypoglycemia, and one of them (via its own receptor) will do it even without an insulin receptor present (Di Cola 1997).
There is also the mitogenic question that sits under all IGF-1 signaling. IGF-1R activation is a proliferative, anti-apoptotic signal, which is precisely why elevated IGF-1 tone is studied in the context of cancer risk. A more potent, less-buffered, binding-protein-independent agonist is not obviously the thing you want more of if you are thinking past this training block. That concern applies to both LR3 and DES.
Bottom line: LR3 versus DES is a duration-versus-local-potency trade on paper, but both sides of that trade are built on preclinical data and marketing, not human outcomes. The most defensible read is that neither has earned a place in human use, and the hypoglycemia and mitogenic risks are real while the performance benefits remain hypothetical.
If you are sourcing for research
For laboratory and research applications, both analogs are available from Ascension Peptides with 50% off using code ENHANCED. Purity and identity matter more here than with almost any other class, given the Mongongu finding that black-market IGF-1 products are routinely oxidized and degraded; a certificate of analysis is not optional. Compare the specifics on the IGF-1 LR3 and IGF-1 DES pages before deciding which, if either, fits your protocol.
This article is for educational and research purposes only. Neither IGF-1 LR3 nor IGF-1 DES is approved for human use, and no controlled human trial has evaluated either for muscle growth, performance, or body composition. Nothing here is medical advice. IGF-1 analogs can cause hypoglycemia and carry theoretical mitogenic and malignancy risks; they should not be self-administered.