At a glance
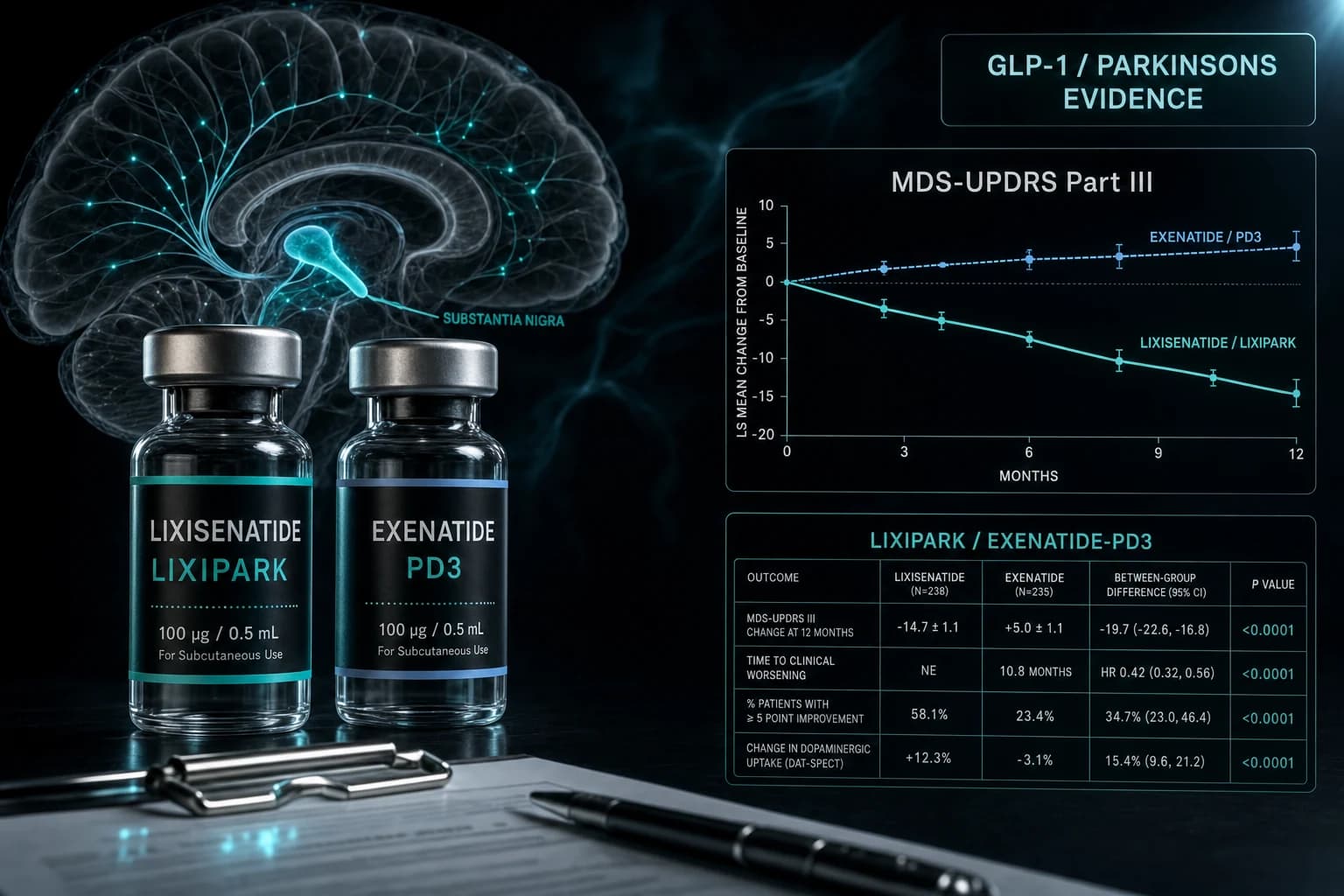
- LIXIPARK Phase 2 cut MDS-UPDRS-III decline by 3.08 points at 12 months (Meissner NEJM 2024, PMID 38598572)
- Exenatide-PD3 Phase 3 found no motor benefit at 96 weeks in 194 patients (Vijiaratnam Lancet 2025, PMID 39919773)
- Athauda 2017 Phase 2 showed 3.5 point OFF-medication MDS-UPDRS-III gain over placebo at 60 weeks (PMID 28781108)
- No GLP-1 receptor agonist is FDA approved for Parkinson's; all clinical use is off-label
- Lixisenatide caused nausea in 46% and vomiting in 13% of LIXIPARK patients
A Phase 2 win and a Phase 3 miss
A French Phase 2 trial in early Parkinson's disease showed that daily lixisenatide slowed motor disability progression by 3.08 MDS-UPDRS-III points over 12 months. A UK Phase 3 trial in moderate Parkinson's, run almost in parallel, found that once-weekly exenatide did nothing. Both drugs are GLP-1 receptor agonists. The reasonable next question is what is happening here, and what should anyone watching this space actually take away from it.
In April 2024, NEJM published the LIXIPARK trial: a French, investigator-initiated, multicenter, double-blind, placebo-controlled Phase 2 study of daily lixisenatide in 156 people with early Parkinson's disease. At 12 months, the change in MDS-UPDRS part III scores was -0.04 points in the lixisenatide arm versus +3.04 points in placebo, a difference of 3.08 points (95% CI 0.86 to 5.30, p=0.007) (Meissner et al. NEJM 2024, PMID 38598572).
Ten months later, in February 2025, The Lancet published Exenatide-PD3: a UK, investigator-initiated, multicenter, double-blind, placebo-controlled Phase 3 trial of once-weekly exenatide 2 mg in 194 people with moderate Parkinson's. At 96 weeks, there was no significant difference in MDS-UPDRS part III change between exenatide and placebo. The trial failed its primary endpoint (Vijiaratnam et al. Lancet 2025, PMID 39919773).
Both findings are real. Both are statistically clean. They tell competing stories. This article walks through how a sober reader of the literature should hold both at once.
The four randomized trials, side by side
Before any narrative, here is the complete RCT record for GLP-1 receptor agonists in Parkinson's disease as of mid-2026:
| Trial | Year | Drug | Phase | n | Population | Duration | Primary endpoint |
|---|---|---|---|---|---|---|---|
| Aviles-Olmos | 2013 | Exenatide 5-10 mcg twice daily | Single-blind, open-label control | 45 | Moderate PD | 12 months treatment + 2 month washout | +4.9 point MDS-UPDRS difference at 12 months (p=0.037) |
| Athauda (Exenatide-PD2) | 2017 | Exenatide 2 mg once weekly | Double-blind, placebo Phase 2 | 62 | Moderate PD | 48 weeks + 12 week washout | +3.5 point MDS-UPDRS-III difference at 60 weeks OFF (p=0.0318) |
| Meissner (LIXIPARK) | 2024 | Lixisenatide 20 mcg daily | Double-blind, placebo Phase 2 | 156 | Early PD (under 3 years), no motor complications | 12 months + 2 month washout | +3.08 point MDS-UPDRS-III difference at 12 months ON (p=0.007) |
| Vijiaratnam (Exenatide-PD3) | 2025 | Exenatide 2 mg once weekly | Double-blind, placebo Phase 3 | 194 | Moderate PD | 96 weeks | No significant MDS-UPDRS-III difference; failed primary endpoint |
Three positives. One negative. The negative is the one that mattered most: the only Phase 3, the longest in duration, and the largest exenatide trial on record.
What Aviles-Olmos 2013 actually established
This was the proof-of-concept study. Aviles-Olmos et al. JCI 2013 (PMID 23728174) randomized 45 patients with moderate Parkinson's to subcutaneous exenatide 5 mcg twice daily for one month then 10 mcg twice daily for 11 months, or to act as untreated controls. At 12 months, exenatide-treated patients had a mean improvement of 2.7 points on MDS-UPDRS, compared with a mean decline of 2.2 points in controls, a 4.9-point adjusted mean difference (p=0.037).
The design has an important caveat: it was single-blind, with an open-label control arm. There was no placebo injection. That is the easiest way to bias a motor scale that depends on observer rating.
The signal was real enough to justify the next trial. It was not real enough to call exenatide a disease-modifying agent.
What Athauda 2017 added
Athauda et al. Lancet 2017 (PMID 28781108) is the trial often shorthanded as Exenatide-PD2. It was the first proper double-blind, placebo-controlled Phase 2 of a GLP-1 receptor agonist in Parkinson's. Sixty-two patients with moderate disease were randomized 1:1 to subcutaneous exenatide 2 mg once weekly or matched placebo for 48 weeks, then washed out for 12 weeks. The primary endpoint was MDS-UPDRS part III at 60 weeks, OFF medication.
Off-medication MDS-UPDRS-III scores improved by 1.0 point in the exenatide group and worsened by 2.1 points in the placebo group: an adjusted difference of -3.5 points (95% CI -6.7 to -0.3, p=0.0318).
This is the result that did most of the work convincing the field that GLP-1 receptor agonists deserved a Phase 3. The OFF-medication design matters: it reduces the chance that any benefit was a symptomatic dopaminergic effect rather than something disease-modifying. But the trial was still small (n=62) and short (60 weeks). The magnitude was modest. The confidence interval almost crossed zero.
What LIXIPARK showed
Meissner et al. NEJM 2024 (PMID 38598572) was the first published Phase 2 to use a different GLP-1 receptor agonist than exenatide. Lixisenatide is a once-daily, short-acting GLP-1 receptor agonist that was marketed as Adlyxin and Lyxumia for type 2 diabetes (Sanofi has since discontinued it in the US market). The trial recruited 156 people with Parkinson's diagnosed less than 3 years earlier, on stable dopaminergic medication, and without motor complications.
Patients were randomized 1:1 to subcutaneous lixisenatide 20 mcg daily or placebo for 12 months, followed by a 2-month washout. The primary endpoint was the change in MDS-UPDRS-III at 12 months in the ON-medication state.
Results: -0.04 points in the lixisenatide arm versus +3.04 points in placebo, difference 3.08 points (95% CI 0.86 to 5.30, p=0.007). After the 2-month washout, the between-group difference shrank to 1.6 points and was no longer statistically significant.
Tolerability was the limiting factor. Nausea occurred in 46% of lixisenatide recipients and vomiting in 13%. Twelve patients in the lixisenatide arm reduced their dose because of side effects.
A few items the headline coverage often missed:
- The endpoint was assessed in the ON state, not OFF. That is a meaningfully different signal than Athauda 2017.
- The washout result eroded the difference. A truly disease-modifying agent would be expected to leave a more durable footprint.
- LIXIPARK was a Phase 2. The benefit needs Phase 3 confirmation before "lixisenatide slows Parkinson's" is a defensible statement.
What Exenatide-PD3 showed
Vijiaratnam et al. Lancet 2025 (PMID 39919773) is the trial that reset expectations. Exenatide-PD3 was a UK-wide, multicenter, double-blind, placebo-controlled Phase 3 of once-weekly subcutaneous exenatide 2 mg versus placebo in 194 people with moderate Parkinson's, run over 96 weeks with no washout phase before primary analysis.
The primary endpoint was the change in MDS-UPDRS part III in the OFF-medication state at 96 weeks. The result: no significant difference between exenatide and placebo. The drug also showed no benefit on secondary outcomes including non-motor symptoms, quality of life, and other Parkinson's severity measures. Exenatide was safe and well tolerated.
So why did Phase 2 land and Phase 3 not? Several explanations are on the table. None has been confirmed:
- Brain penetration. Cerebrospinal fluid analysis from the Phase 2 program suggested exenatide reaches the brain only in low concentrations. If exposure at the target tissue is sub-therapeutic, no amount of trial size will rescue the effect.
- Population. Phase 2 enrolled patients with moderate, established PD. Phase 3 used similar inclusion criteria. Lixisenatide enrolled patients with shorter disease duration and no motor complications. The disease-modifying window may close earlier than the exenatide trials targeted.
- Endpoint state. Both Athauda 2017 and Exenatide-PD3 used OFF-medication MDS-UPDRS-III, so endpoint definition does not explain the divergence between those two exenatide trials.
- Statistical fluke. The Phase 2 result had a wide confidence interval and a marginal p value. Regression to the null in Phase 3 is one of the most common failure modes for promising small trials.
Bottom line: The most parsimonious read of the literature is that GLP-1 receptor agonists may have a real, modest motor benefit in early Parkinson's but no measurable disease-modifying effect in moderate Parkinson's. The lixisenatide signal is interesting but unconfirmed and includes a substantial nausea and vomiting cost.
What the animal data say (and do not)
Preclinical work has been consistent. Multiple labs have reported neuroprotective effects of GLP-1 receptor agonists in toxin (MPTP, 6-OHDA) and genetic mouse models of PD, with reduced dopaminergic neuron loss, improved motor performance, and lower alpha-synuclein burden. The semaglutide-specific work, Zhang et al. 2019 (PMID 30741689), showed that semaglutide rescued tyrosine hydroxylase expression and reduced alpha-synuclein in the chronic MPTP model.
Two important limits:
- Mouse MPTP models are acute and stereotyped. They are useful for screening neuroprotection. They are bad for predicting whether a drug will modify a slow, complex human disease.
- Animal-model success has translated poorly to clinical disease modification across the entire PD pipeline. Anti-alpha-synuclein antibodies, kinase inhibitors, and isradipine all crashed in Phase 2 or 3. Exenatide-PD3 is the most recent entry to that ledger.
The 2020 Cochrane review on GLP-1 receptor agonists for PD (PMID 32700772) reached a similar caution before the Phase 3 read out, finding only low-certainty evidence that exenatide improves motor function. The Phase 3 result is consistent with that low-certainty grade.
What is still in flight
Several GLP-1 trials in PD are ongoing or recruiting in 2026:
- Semaglutide. A long-running Phase 2 of subcutaneous semaglutide once weekly with motor outcomes plus DAT-SPECT imaging has been registered but has not yet published a primary readout.
- Liraglutide. Smaller academic trials have been recruiting in the US and Europe.
- Combinations and dual agonists. No registered Phase 2 yet for tirzepatide or retatrutide in PD as of this writing.
The next inflection point is the semaglutide Phase 2 readout. Semaglutide is the GLP-1 receptor agonist most often cited as having stronger blood-brain barrier penetration than exenatide and a longer half-life, which is the most-cited reason to expect a different result. Whether that translates to a clean motor signal is what the next trial cycle will tell.
For broader context on what the GLP-1 class is being repurposed for, our stopping GLP-1s research read and the semaglutide alcohol cravings research read cover two of the better-evidenced non-metabolic uses.
How to read this if you or a family member has PD
A few honest framings, in plain English:
- No GLP-1 receptor agonist is approved for Parkinson's disease anywhere in the world. The FDA has approved no PD indication for semaglutide, tirzepatide, exenatide, or lixisenatide. Use in PD is off-label. Unsupervised use carries the GI side effect profile (nausea, vomiting, gastroparesis), pancreatitis risk, and the lean-mass loss footprint we cover in GLP-1 muscle loss research.
- The strongest signal is in early disease. LIXIPARK enrolled people within 3 years of diagnosis. The exenatide trials enrolled moderate disease, and the Phase 3 was negative. If a clinician is going to consider a GLP-1 receptor agonist off-label in PD, the most defensible scenario based on current data is early disease.
- The benefit, even where seen, is modest. A 3-point MDS-UPDRS-III difference at 12 months is real but small. It is not the kind of effect size that reverses or arrests disease.
- Tolerability is a real cost. Lixisenatide's 46% nausea rate is high enough that "well tolerated" overstates the case for many patients. Slow titration helps but does not eliminate the issue.
- Brain-penetrant GLP-1 receptor agonists are the next test. Until a more CNS-active GLP-1 agent reads out positive in Phase 3, the field has no compound with confirmed disease-modifying activity in PD.
Tip: If you are weighing this with a movement disorder neurologist, the most useful question is not "should I take Ozempic for my Parkinson's?" but "do you have an opinion on whether the LIXIPARK signal is large enough to justify off-label use in early disease?" That conversation will tell you more than any forum thread.
How this fits with the rest of the GLP-1 evidence base
The class is good at what it was built to do: weight loss and glycemic control. The most rigorous benefits sit there. For the comparative head-to-head between the three approved compounds for weight loss, see retatrutide vs tirzepatide vs semaglutide 2026 and tirzepatide vs semaglutide 2026.
The class also has emerging signals in cardiometabolic comorbidities (heart failure, sleep apnea, MASH) where the Phase 3 evidence is stronger than the Parkinson's data. See tirzepatide for HFpEF after SUMMIT, tirzepatide for sleep apnea after SURMOUNT-OSA, and semaglutide for MASH after ESSENCE for the comorbidity reads.
The neurological repositioning story is at an earlier maturity than any of those. Parkinson's is the most-studied non-metabolic indication in this category. Alzheimer's, alcohol use disorder, and substance use disorder all have smaller signals so far, with semaglutide and alcohol cravings being the cleanest of the non-PD reads.
The honest summary
Three positive trials and one negative trial. The negative is the largest, longest, most rigorous study of the most-tested compound. The positives are smaller, shorter, and one (Aviles-Olmos 2013) was not blinded. The lixisenatide signal in early disease is the most credible positive on the board, and it is one Phase 2 trial.
What this means in practice:
- As clinical evidence: GLP-1 receptor agonists are not established as disease-modifying in Parkinson's disease. Anyone who tells you otherwise is overselling the data.
- As research interest: The class still deserves a Phase 3 of a brain-penetrant GLP-1 agonist in early PD before the file is closed.
- As a personal decision: Off-label use in early disease, under specialist supervision, is defensible if the patient understands they are betting on an unconfirmed Phase 2 signal and accepting the GI cost.
That is what the trials show. Not what the headlines show.
Further reading
- GLP-1 muscle loss: what the research shows in 2026
- Stopping GLP-1s: weight regain research
- Semaglutide for alcohol cravings: 2025 research
- Semaglutide for MASH: ESSENCE Phase 3 evidence
- Tirzepatide for HFpEF: SUMMIT trial evidence
- Retatrutide vs tirzepatide vs semaglutide 2026
- Semaglutide compound page
- Tirzepatide compound page
- Reconstitution calculator
References
- Aviles-Olmos I, et al. Exenatide and the treatment of patients with Parkinson's disease. J Clin Invest. 2013;123(6):2730-2736. PMID 23728174
- Athauda D, et al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390(10103):1664-1675. PMID 28781108
- Mulvaney CA, et al. GLP-1 receptor agonists for Parkinson's disease. Cochrane Database Syst Rev. 2020;7:CD012990. PMID 32700772
- Zhang L, et al. Semaglutide is neuroprotective and reduces alpha-synuclein levels in the chronic MPTP mouse model of Parkinson's disease. J Parkinsons Dis. 2019;9(1):157-171. PMID 30741689
- Meissner WG, et al. Trial of lixisenatide in early Parkinson's disease. N Engl J Med. 2024;390(13):1176-1185. PMID 38598572
- Vijiaratnam N, et al. Exenatide once a week versus placebo as a potential disease-modifying treatment for people with Parkinson's disease in the UK: a phase 3, multicentre, double-blind, parallel-group, randomised, placebo-controlled trial. Lancet. 2025;405(10481):627-636. PMID 39919773
This article is for educational and research purposes only. None of the content above constitutes medical advice. No GLP-1 receptor agonist is FDA approved for Parkinson's disease. Decisions about Parkinson's disease management belong with the patient and their movement disorder neurologist.